

HPRA Registration in Ireland: A Step-by-Step Guide for Medical Device Manufacturers
If you manufacture medical devices or in vitro diagnostic (IVD) devices and want to place them on the Irish market, you will need to register with the Health Products Regulatory Authority (HPRA).
The HPRA is Ireland’s national competent authority for medical devices. It operates within the framework of the EU Medical Device Regulation (EU MDR 2017/745) and the EU IVD Regulation (EU IVDR 2017/746), and is responsible for market surveillance, vigilance, and registration of devices sold in Ireland.
This guide walks through the HPRA registration process step by step. From confirming your device classification through to maintaining ongoing compliance once your device is on the market.
Why Ireland matters for EU market access
Ireland is an important market in its own right, but for many non-EU manufacturers it also acts as a gateway to the wider European single market. Because Ireland is an EU member state, manufacturers and authorised representatives based there operate within the EU regulatory framework. It is CE marking — combined with registration on EUDAMED — that allows a device to circulate freely across all 27 EU member states, not registration with the HPRA specifically. The HPRA is the relevant competent authority for devices placed on the Irish market and acts as the primary regulatory contact for manufacturers and authorised representatives established in Ireland.
Ireland is also one of the most commonly chosen jurisdictions for EU Authorised Representative services, particularly among US and UK-based device manufacturers. For companies based outside the EU, including those in the United States, UK, Australia, and Asia, appointing an EU Authorised Representative in Ireland can simplify the compliance process. Instead of dealing with multiple authorities, manufacturers work primarily with the HPRA while maintaining access to the full EU market.
The HPRA registration process: step by step
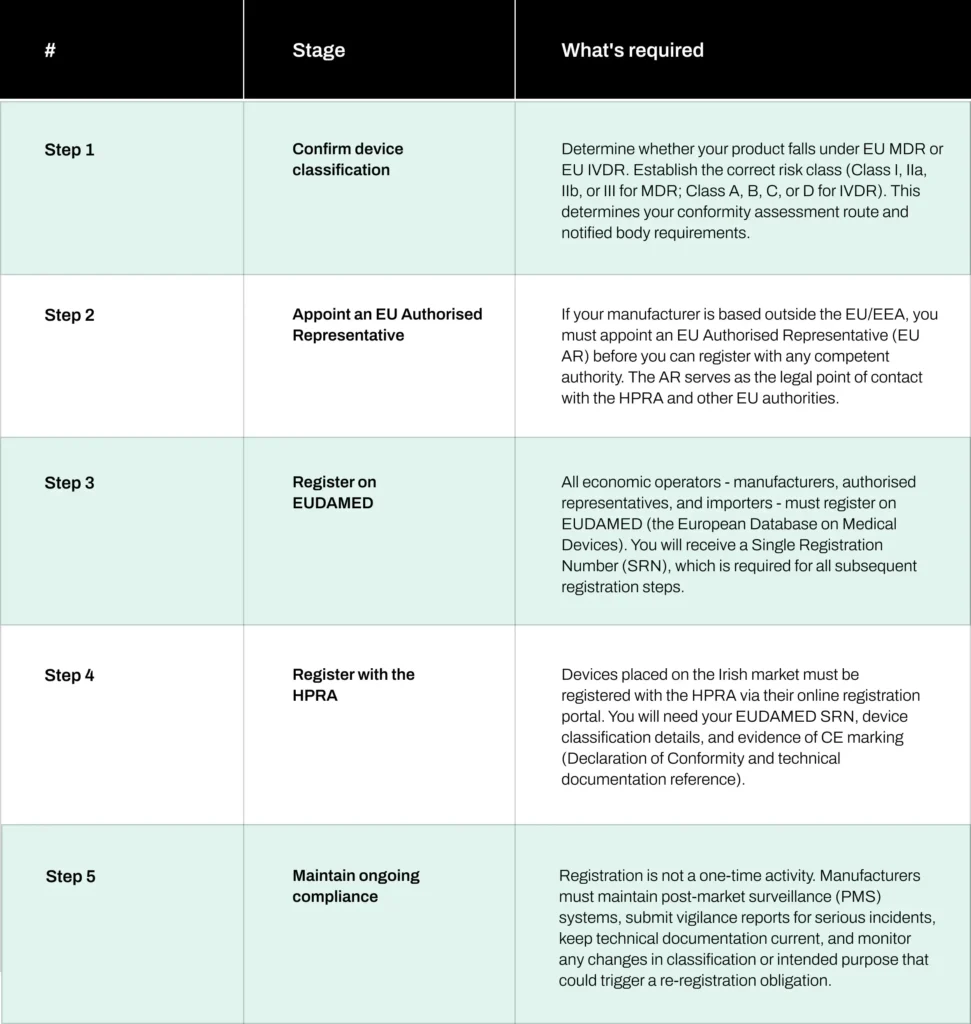
A closer look at each step
Step 1: Device classification
Before starting the registration process, you need to confirm the correct classification for your device. Under the EU MDR, devices are classified as Class I, IIa, IIb, or III based on their risk profile and intended purpose. Under the EU IVDR, classifications range from Class A, which carries the lowest risk, to Class D, which carries the highest.
Classification is not always straightforward. The rules are defined in Annex VIII of both the EU MDR and EU IVDR, and applying them correctly requires careful assessment of the device’s intended purpose, mode of action, and level of patient contact. Incorrect classification can lead manufacturers down the wrong conformity assessment route, resulting in delays, unnecessary costs, and potential regulatory action.
• Class I devices that are non-sterile, have no measuring function, and are not reusable surgical instruments can self-declare conformity without a notified body.
• Class IIa, IIb, and III devices require involvement of a notified body for conformity assessment.
• There are three Class I sub-categories that do require notified body involvement for those specific aspects: sterile Class I devices (Is), Class I devices with a measuring function (Im), and reusable surgical instruments (Ir). If your Class I device falls into any of these sub-categories, you will need to involve a notified body for the relevant part of your conformity assessment even though the overall device remains Class I.
Step 2: EU Authorised Representative
Non-EU manufacturers must appoint an EU Authorised Representative before registering a device in any EU member state. The EU AR is listed on the device label and included in all regulatory documentation. They act as the legal point of contact between the manufacturer and EU competent authorities, including the HPRA.
The EU AR’s responsibilities include registering in EUDAMED on behalf of the manufacturer, cooperating with competent authorities on post-market surveillance and vigilance activities, and maintaining copies of the Declaration of Conformity and technical documentation.
Appointing an EU Authorised Representative is a legal requirement under Article 11 of both the EU MDR and EU IVDR. Devices without an appointed EU AR cannot legally be placed on the EU market.
Step 3: EUDAMED registration
EUDAMED is the European Commission’s central database for medical devices. Registration in EUDAMED is mandatory for manufacturers, authorised representatives, and importers under both the EU MDR and EU IVDR.
Once registered, you receive a Single Registration Number (SRN). This number is required before you can register devices with national competent authorities, including the HPRA. Without an SRN, you cannot move forward with the registration process.
From 28 May 2026, four EUDAMED modules become mandatory:
- Actor Registration — mandatory from 28 May 2026 with no transition period. If you are a manufacturer, authorised representative, importer, or system and procedure pack producer and have not yet completed actor registration, this must be done immediately.
- UDI/Device Registration — mandatory from 28 May 2026 for all MDR/IVDR devices placed on the market on or after that date. Devices already on the market before 28 May 2026 (legacy and MDR/IVDR devices) have a 12-month transition period, with a deadline of 28 November 2026.
- Notified Bodies and Certificates — mandatory from 28 May 2026, with an 18-month transition period to register information for MDR and IVDR certificates issued before the mandatory use date. Deadline: 28 May 2027.
- Market Surveillance — mandatory from 28 May 2026 with no transition period.
If your actor registration is not yet complete, this is the immediate priority. Devices placed on the market on or after 28 May 2026 must be registered in the UDI/Device module before the first unit is placed on the market — there is no transition period for new devices.
Your EU Authorised Representative can help manage the EUDAMED registration process and ensure all required modules are completed correctly and on time.
Step 4: HPRA registration
With your SRN in hand, you can proceed to register your device with the HPRA via their online registration portal. You will be required to provide:
• Your EUDAMED Single Registration Number (SRN)
• Device classification and intended purpose
• CE marking documentation (Declaration of Conformity)
• Technical file reference and notified body certificate (where applicable)
• Contact details for the EU Authorised Representative
The HPRA may request additional information or documentation. Response times and registration timelines can vary depending on device class and submission quality.
Step 5: Ongoing compliance
Registration with the HPRA is not a one-time milestone. It is the start of an ongoing compliance obligation. Under EU MDR and EU IVDR, manufacturers must:
• Operate a post-market surveillance (PMS) system. Class I manufacturers are required to maintain a PMS report. Manufacturers of Class IIa, IIb, and III devices are additionally required to produce Periodic Safety Update Reports (PSURs) — a more structured and regularly updated document with defined update frequencies depending on device class.
• Report serious incidents and field safety corrective actions (FSCAs) to the HPRA via EUDAMED
• Keep technical documentation up to date and ensure it reflects any changes to the device
• Notify the HPRA and update EUDAMED if the device classification, intended purpose, or responsible parties change
Failure to meet these ongoing obligations can result in market withdrawal or enforcement action by the HPRA or other national competent authorities. Separately, a notified body may suspend or withdraw a CE certificate where surveillance audits of the manufacturer’s quality management system identify non-conformities.This is a parallel enforcement route, distinct from competent authority action.
How Euverify can help
Euverify provides end-to-end medical device compliance support for manufacturers seeking to place devices on the EU and UK markets. Our services include:
• EU Authorised Representative services (Ireland-based, covering all 27 EU member states)
• EUDAMED registration and ongoing management
• HPRA registration support
• MHRA registration and UK Responsible Person services
• Classification advice and conformity assessment guidance
• Post-market surveillance and vigilance support
Whether you are registering your first device or managing a large portfolio across multiple markets, our team has the regulatory expertise to guide you through every step of the process.
Key Takeaways
Once your device is CE marked and registered on EUDAMED, it can be placed on the market across all 27 EU member states. However, manufacturers still need to meet any additional national registration requirements that apply, including those enforced by the HPRA in Ireland.
Preparing the correct documentation in advance, including your EUDAMED SRN, Declaration of Conformity, and technical files, can help avoid unnecessary delays during the registration process. Choosing an EU Authorised Representative based in Ireland also gives manufacturers a clear regulatory contact point through the HPRA, which many companies find practical due to Ireland’s established life sciences sector and English-language regulatory environment.
Ready to register your device in Ireland?
Contact Euverify today to discuss your requirements. Our team of regulatory specialists will guide you through every stage of the HPRA registration process and ensure your device reaches the EU market compliantly and efficiently.









